Autophagy Activation: Timing and Triggers in Humans
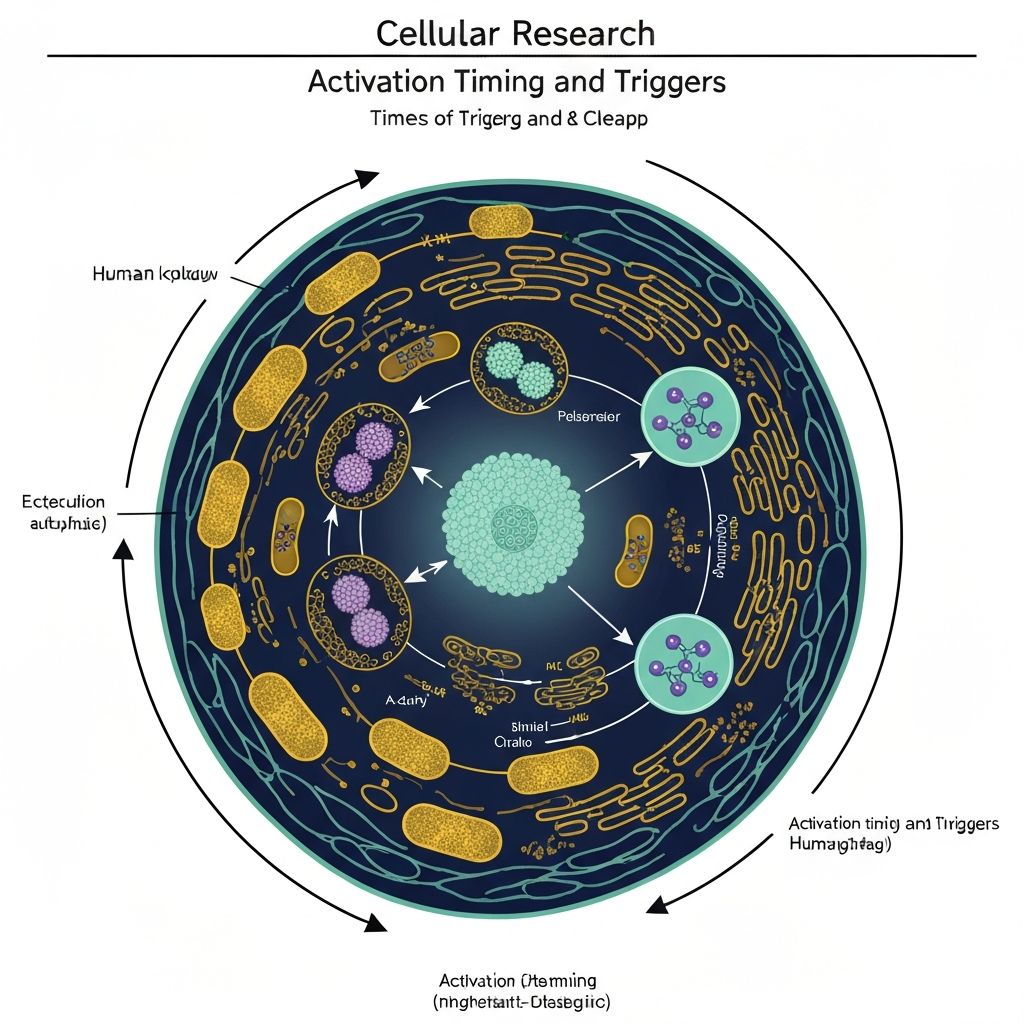
Autophagy: Cellular Degradation and Renewal
Autophagy, derived from Greek "auto" (self) and "phagy" (eating), describes catabolic cellular processes wherein cells engulf and degrade their own intracellular components. This process serves fundamental housekeeping functions, removing damaged organelles, protein aggregates, and dysfunctional cellular material.
Three primary autophagic pathways exist: macroautophagy (large-scale engulfment of cytoplasmic contents), microautophagy (direct lysosomal engulfment of cytoplasm), and chaperone-mediated autophagy (selective protein transport into lysosomes). Macroautophagy represents the predominant pathway and primary focus of nutritional autophagy research.
Basal autophagy occurs constitutively at relatively low rates, serving continuous cellular maintenance functions. However, nutrient deprivation, particularly extended fasting, upregulates autophagic flux substantially, creating heightened cellular degradation and recycling activity. This fasting-induced autophagy represents an adaptive response mobilising intracellular resources during nutrient scarcity.
Molecular Triggers of Autophagy Activation
mTOR Pathway Inhibition: The mechanistic target of rapamycin (mTOR) represents a central nutrient-sensing kinase. When amino acid concentrations are adequate and energy status is replete, mTOR maintains active phosphorylation, suppressing autophagy. Conversely, amino acid depletion (characteristic of fasting) inactivates mTOR, removing autophagy suppression and permitting activation.
AMPK Activation: The AMP-activated protein kinase (AMPK) functions as an energy sensor, becoming activated when cellular energy charge declines (elevated AMP:ATP ratio). AMPK phosphorylates and inactivates mTOR, promoting autophagy during energy-depleted conditions. Fasting-induced energy depletion robustly activates AMPK, driving autophagy acceleration.
FoxO Transcription Factors: Forkhead box O transcription factors, particularly FoxO3, transactivate autophagy-related genes (ATG genes) encoding proteins essential for autophagic machinery assembly. Fasting-induced signalling cascades activate FoxO translocation into the nucleus, enhancing ATG gene transcription and increasing autophagic capacity.
ER Stress Response: Endoplasmic reticulum (ER) stress from accumulation of misfolded proteins triggers the unfolded protein response (UPR). Certain UPR branches activate autophagy as a compensatory mechanism, removing protein aggregates and reducing ER proteotoxic stress.
Phases of Macroautophagic Flux
Autophagosome Formation: The process initiates with formation of a double-membrane vesicle termed a phagophore or isolation membrane, which progressively expands to engulf cytoplasmic contents including organelles, protein aggregates, and ribosomes. This structure seals into a completed autophagosome, a double-membrane vesicle containing the cytoplasmic cargo.
Lysosomal Fusion: The completed autophagosome docks at lysosomal surfaces via SNARE protein interactions, and the outer autophagosomal membrane fuses with the lysosomal membrane. This fusion permits delivery of the autophagosomal content into the lysosomal lumen.
Degradation and Recycling: Lysosomal hydrolases—including proteases, lipases, and nucleases—degrade the autophagosomal content into constituent molecular components. Metabolic products (amino acids, fatty acids, nucleotides) are recycled for biosynthetic and energy-production purposes, or exported into cytoplasm for utilisation by other cellular processes.
This coordinated degradation-recycling process permits intracellular substrate mobilisation during nutrient scarcity, providing alternative fuel sources when exogenous nutrient availability is limited.
Temporal Dynamics of Autophagy in Humans
Basal Autophagy (Fed State): Continuous low-level autophagy maintains cellular homeostasis under normal nutritional conditions. Circulating amino acids and glucose maintain mTOR activity, restraining autophagic flux. This basal autophagy provides essential housekeeping, selectively removing damaged or long-lived proteins and dysfunctional organelles.
Early Fasting (0-12 hours): As postprandial nutrient absorption concludes and blood glucose declines, mTOR begins to downregulate. Autophagic flux increases modestly above basal levels, though acceleration remains gradual. Emerging energy depletion activates AMPK and initiates FoxO-mediated ATG gene upregulation.
Substantial Fasting (24-48 hours): Extended amino acid deprivation and pronounced energy depletion strongly inhibit mTOR and robustly activate AMPK. Autophagic flux reaches substantially elevated levels, with marked upregulation of autophagosome formation and clearance of damaged organelles. Circulating biomarkers of autophagy (putrescine, spermidine) elevate detectably.
Prolonged Fasting (48+ hours): Maximal autophagic activation characterises this phase, with quantitatively significant intracellular degradation and recycling. This intense renovation phase represents a proposed mechanism linking extended fasting to cellular rejuvenation and longevity enhancement in experimental animal models.
Selective Autophagy: Targeting Dysfunctional Components
Beyond non-selective macroautophagy, cells employ selective autophagic pathways targeting specific cargo:
Mitophagy: Selective autophagy of dysfunctional mitochondria. Depolarised mitochondria (membrane potential loss) or those experiencing oxidative stress undergo selective sequestration and degradation via mitophagy. This selective removal prevents accumulation of energy-producing organelles exhibiting dysfunction, maintaining mitochondrial quality control.
Ribophagy: Selective autophagy of ribosomes and ribosomal proteins. During fasting-induced protein synthesis suppression, ribosomal components undergo selective autophagy and degradation, providing amino acids for critical biosynthetic processes or energy production.
ER-Phagy: Selective autophagy of endoplasmic reticulum segments. Abnormal ER regions or those laden with misfolded proteins undergo selective removal via autophagy, reducing proteotoxic burden.
These selective mechanisms ensure autophagy preferentially removes damaged or dispensable components rather than engulfing cellular contents non-discriminately, maintaining essential cellular infrastructure whilst eliminating harmful accumulations.
Human Research Evidence on Autophagy
Direct measurement of autophagic flux in living human tissues remains technically challenging, as autophagy primarily occurs within intracellular vesicular compartments inaccessible to non-invasive investigation. Consequently, human autophagy research employs indirect biomarker approaches:
Plasma Biomarkers: Circulating amino acids and polyamines (putrescine, spermidine) elevated during fasting reflect increased proteolysis and autophagy. Plasma citrulline concentrations, a marker of intestinal epithelial cell turnover, elevate during fasting, reflecting enhanced gut autophagy.
Cellular Models: Human fibroblasts and other cell cultures derived from human tissues demonstrate robust fasting-induced autophagy in vitro. These models permit direct visualisation of autophagosome formation and flux measurement but lack the physiological complexity of intact organisms.
Mechanistic Extrapolation: Fasting-induced signalling pathway activation documented in humans (mTOR inhibition, AMPK activation, FoxO translocation) mechanistically supports autophagy enhancement, though direct flux confirmation remains indirect.
Current scientific understanding indicates that extended human fasting (24+ hours) robustly activates autophagic signalling and induces molecular conditions favouring autophagy; however, quantitative autophagy magnitude in humans remains imperfectly characterised.
Continue Exploring
Learn about energy substrate switching during fasting:
Read the full scientific explanation